Anova
Post-Hoc analysis Solutions
Packages
Creating variables and working with missing values
It’s important to be able to check for, and remove missing values from a data set. You will not be tested on this, but let’s practice this together! Comment through the code below:
penguins |>
mutate(missing = is.na(bill_length_mm)) |> #make a new variable called missing
filter(missing == TRUE) # only select rows that have TRUE in the missing variable# A tibble: 2 × 9
species island bill_length_mm bill_depth_mm flipper_length_mm body_mass_g
<fct> <fct> <dbl> <dbl> <int> <int>
1 Adelie Torgersen NA NA NA NA
2 Gentoo Biscoe NA NA NA NA
# ℹ 3 more variables: sex <fct>, year <int>, missing <lgl>Saving a new data set
Comment through the code below
Note: There are functions like drop_na(): which removes rows containing any NA values. These can be dangerous…
Anova
Now, let’s conduct an ANOVA test between species and bill length. First, let’s check our assumptions.
Normality
The sample = argument within the aes() (aesthetic) function in ggplot2 orders our data on the y-axis. stat_qq_line() maps the theoretical quantiles to our plot.
penguins |>
ggplot(
aes(sample = bill_length_mm)) +
stat_qq() +
stat_qq_line() +
facet_wrap(~ species) + # Replace 'Species' with your grouping variable
labs(title = "QQ Plots of Bill Length by Species",
x = "Theoretical Quantiles (what we would expect to see from a Z)",
y = "Ordered data") +
theme_minimal()Warning: Removed 2 rows containing non-finite outside the scale range
(`stat_qq()`).Warning: Removed 2 rows containing non-finite outside the scale range
(`stat_qq_line()`).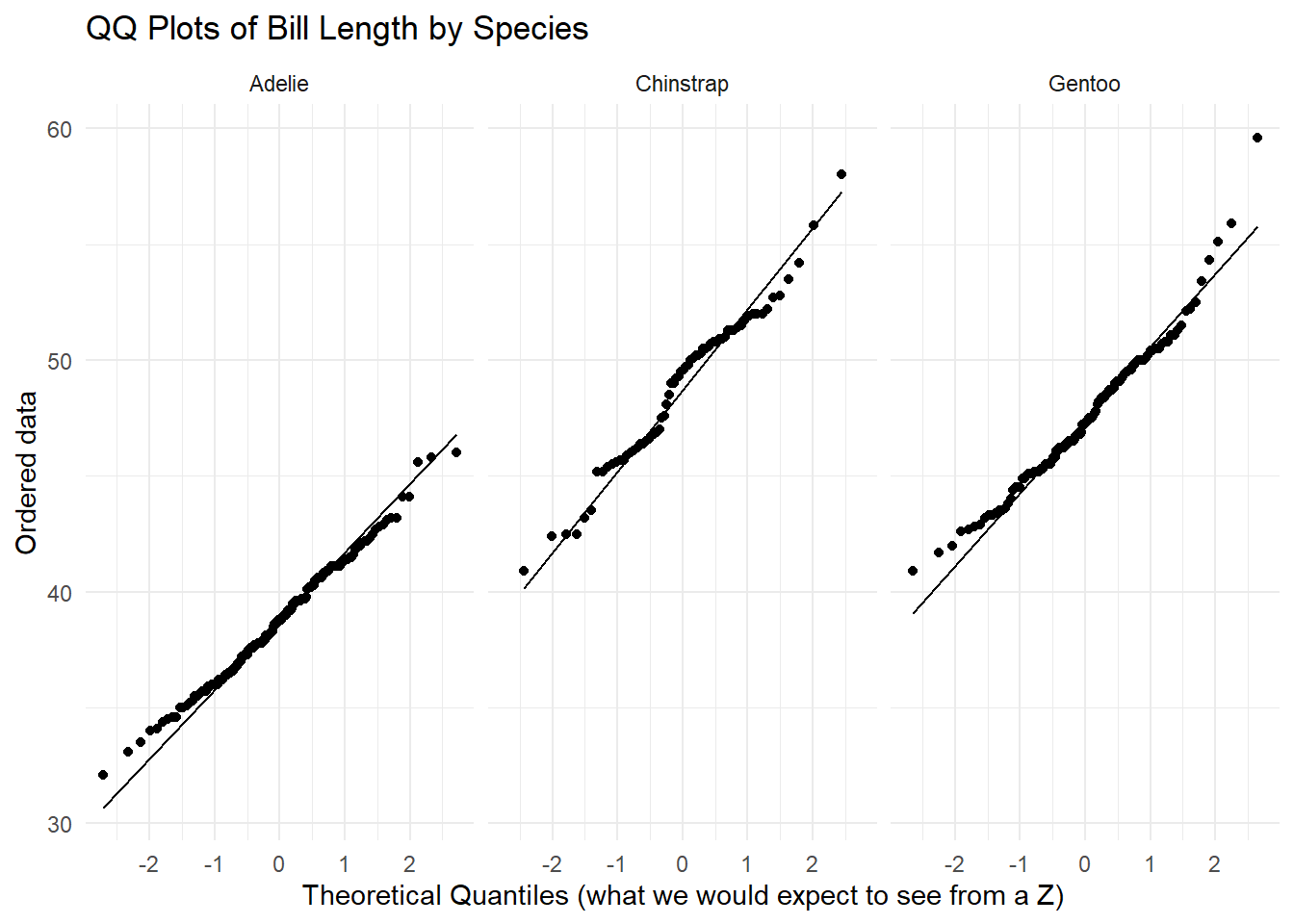
For each of the three species, we see a linear relationship between our ordered data and what we would expect to see from a normal distribution. There is not enough evidence to suggest that the normality assumption is violated.
Equal Variance
Use the appropriate R code below to justify the equal variance assumption. Note: The general rule is the largest standard deviation should not be two times the smallest.
Anova
Fill in the blanks below to conduct your test for anova.
Df Sum Sq Mean Sq F value Pr(>F)
species 2 7194 3597 410.6 <2e-16 ***
Residuals 339 2970 9
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1We can start working with values when the table is a tibble. We can use the tidy() function from the broom package.
tidy(anova_model)# A tibble: 2 × 6
term df sumsq meansq statistic p.value
<chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 species 2 7194. 3597. 411. 2.69e-91
2 Residuals 339 2970. 8.76 NA NA tb <- tidy(anova_model) # save the tibble as tb
tb_species <- tb |>
filter(term == "species")
tb_species$statistic[1] 410.6003tb_species$p.value[1] 2.694614e-91You can write in-line code by putting backticks (instead of ” “) around the”r your code here”. Put the R code you want to execute in-line below within your sentence!
Our statistic is 410.600255
Our p-value is rtb_species$p.value`
Tukeys
To conduct Tukeys HSD, run the code below.
TukeyHSD(anova_model) Tukey multiple comparisons of means
95% family-wise confidence level
Fit: aov(formula = bill_length_mm ~ species, data = peng_filter)
$species
diff lwr upr p adj
Chinstrap-Adelie 10.042433 9.024859 11.0600064 0.0000000
Gentoo-Adelie 8.713487 7.867194 9.5597807 0.0000000
Gentoo-Chinstrap -1.328945 -2.381868 -0.2760231 0.0088993